Na początku tego miesiąca miało miejsce pewne poruszenie w związku z opublikowaniem artykułu (na PrePrint[1]), zatytułowanego „Poważne niepożądane zdarzenia o szczególnym znaczeniu po szczepieniu mRNA w badaniach z randomizacją” („Serious Adverse Events of Special Interest Following mRNA Vaccination in Randomized Trials”).
Do opisu wyników użyto słów takich jak „badanie przełomowe”. Brzmi to dość znacząco i oczywiście przyciągnęło moją uwagę. Skoro redaktor pisma BMJ [dawnej: British Medical Journal], dr Peter Doshi, jest głównym autorem, to rzeczywiście takim badaniem przełomowym może być! Dr Doshi cieszy się zasłużoną reputacją osoby mówiącej niewygodne prawdy. Przyjrzyjmy się więc temu.
Oto najważniejsze wyniki ze streszczenia:
- Szczepionki mRNA COVID-19 firmy Pfizer i Moderna były związane ze zwiększonym ryzykiem poważnych zdarzeń niepożądanych o szczególnym znaczeniu, z bezwzględnym wzrostem ryzyka o 10,1 i 15,1 na 10 000 zaszczepionych w porównaniu z placebo, wynoszącym odpowiednio 17,6 i 42,2 (95% CI -0,4 do 20,6 i -3,6 do 33,8).
- Łącznie szczepionki mRNA wiązały się z bezwzględnym zwiększeniem ryzyka wystąpienia poważnych zdarzeń niepożądanych o szczególnym znaczeniu o 12,5 na 10 000 (95% CI 2,1 do 22,9).
- Nadmierne ryzyko wystąpienia poważnych zdarzeń niepożądanych o szczególnym znaczeniu przewyższało zmniejszenie ryzyka hospitalizacji z powodu szczepionki COVID-19 w porównaniu z grupą placebo w obu badaniach firmy Pfizer i Moderna (odpowiednio 2,3 i 6,4 na 10 000 uczestników).
A w części dyskusyjnej streszczenia:
„Stwierdzone w naszym badaniu nadmierne ryzyko poważnych zdarzeń niepożądanych wskazuje na potrzebę przeprowadzenia formalnych analiz stosunku korzyści do szkód, zwłaszcza stratyfikowanych według ryzyka wystąpienia poważnych następstw stosowania COVID-19, takich jak hospitalizacja lub zgon”.
Na pierwszy rzut oka nagłówki wyglądają całkiem poważnie. Jednak część dyskusyjna powinna nas ostrzec, że autorzy zachowują ostrożność. Nie sygnalizują oni, że chodzi o bezczelne kłamstwo.”
Co zatem tak naprawdę dzieje się w tym przypadku? Aby to zrozumieć, warto zacząć od tego niezwykle przejrzystego i dokładnego podsumowania wstępnych wyników badania firmy Pfizer, przygotowanego przez kanadyjskie stowarzyszenie COVID Care Alliance (zamieszczenie tego tekstu jest najwyraźniej grzechem, przez który w grudniu ubiegłego roku zostałem wyrzucony z serwisów Twitter i Linked-In, co spowodowało odcięcie mnie od około 600 000 obserwatorów).
Podsumowanie tej analizy i ustaleń w formacie PDF można znaleźć tutaj.
Wniosek jest taki, że badanie fazy 3 firmy Pfizer, które zostało wykorzystane przez NIAID, FDA i CDC do uzasadnienia zezwolenia na zastosowanie [„szczepionki”] w nadzwyczajnych wypadkach[2], jest w zasadzie nic nie wartym[3] badaniem klinicznym, które zostało niewłaściwie wstrzymane na długo przed zbliżeniem się do planowanego okresu obserwacji, nie dostarczyło wystarczająco długiej analizy zdarzeń niepożądanych związanych ze szczepieniem i w którym celowo wyeliminowano grupę kontrolną. W rezultacie, w zasadzie zlikwidowano wszelkie możliwości dotarcia do sedna prawdziwego ryzyka związanego ze szczepieniami mRNA firmy Pfizer. Jeśli chodzi o mniejsze zagrożenia, badanie nie miało odpowiedniej mocy (nie było wystarczająco duże), aby je ocenić.
Do akcji wkroczyła nieustraszona grupa (głównie) starszych badaczy akademickich. Przychodzi na myśl powiedzenie „głupcy wchodzą tam, gdzie anioły boją się chodzić”, ponieważ kwestionowanie przez naukowców przyjętej narracji o szczepionkach stało się niezwykle ryzykowne. Jednak ta nie-tak-głupia grupa odważnie przystąpiła do działania..
Według mnie podejście, jakie przyjęli w tej analizie i raporcie, polegało na podjęciu w dobrej wierze wysiłku przeprowadzenia analizy badań klinicznych fazy 3 (są to rzekomo „duże, końcowe” badania kliniczne przed dopuszczeniem produktu do obrotu), które powinny być przeprowadzone przez firmy Moderna i Pfizer. Zasadniczo chodzi o analizy, które FDA powinna była wykonać sama, a także powinna była zmusić do tego firmy Moderna i Pfizer. Gdyby szef personelu Białego Domu, Mark Meadows, nie wywierał presji na FDA, być może FDA postąpiłaby słusznie. Ale FDA najwyraźniej ugięła się i nie zrobiła tego, co do niej należało, i oto mamy to co mamy.
W tym tkwi sedno sprawy: FDA nie tylko nie wykonała swojego zadania, ale ani FDA, ani Moderna, ani Pfizer nie opublikują danych pierwotnych, co oznacza, że nikt inny też nie może tego zrobić. Jak zauważają w swoim omówieniu autorzy tej ostatniej analizy:
„Należy przeprowadzić systematyczny przegląd i metaanalizę z wykorzystaniem danych dotyczących poszczególnych uczestników, aby odpowiedzieć na pytania dotyczące stosunku szkodliwości do korzyści w różnych podgrupach demograficznych. Aby właściwie ocenić te kwestie, konieczna jest pełna przejrzystość danych z badań klinicznych nad szczepionką COVID-19. Niestety, ponad rok po powszechnym stosowaniu szczepionki COVID-19 dane dotyczące poszczególnych uczestników są nadal niedostępne.”
Doshi i współpracownicy wielokrotnie wzywali do pełnego ujawnienia danych w dwóch wcześniejszych publikacjach[4], ale bezskutecznie. Jeśli więc dane te nie zostaną włączone do nakazanego przez sąd ujawnienia danych[5], analiza przeprowadzona przez nich w obecnym raporcie może być jedyną dobrą, jaką otrzymamy. Więcej informacji na ten temat można znaleźć na stronie:
- Tanveer S, Rowhani-Farid A, Hong K, Jefferson T, Doshi P. Transparency of COVID-19 vaccine trials: decisions without data. BMJ Evid Based Med [Internet]. 2021 Aug 9
- Doshi P, Godlee F, Abbasi K. Covid-19 vaccines and treatments: we must have raw data, now. BMJ [Internet]. 2022 Jan 19;376:o102.
Jak słusznie zauważają dr Doshi i współpracownicy:
„W 2013 r. amerykańskie i europejskie organizacje branżowe poparły wspólne oświadczenie w sprawie udostępniania danych z badań klinicznych, podejmując szereg zobowiązań, które 'uznają znaczenie udostępniania danych z badań klinicznych w interesie pacjentów, opieki zdrowotnej i gospodarki’. W 2015 r. amerykański Instytut Medycyny podobnie poparł korzyści płynące z udostępniania danych z badań klinicznych, podkreślając, że 'weryfikacja i replikacja twierdzeń badaczy’ są niezbędne dla procesu naukowego i zauważając liczne korzyści dla zainteresowanych stron, 'w tym płatników opieki zdrowotnej, a także pacjentów, ich lekarzy i naukowców.’”
Ale „gdyby życzenia były końmi, żebracy jeździliby konno„. Pfizer, Moderna i FDA najwyraźniej nie mają zamiaru wysłuchać próśb starszego redaktora British Medical Journal, chyba że zmuszą ich do tego amerykańskie sądy, a nawet wtedy będą zwlekać tak długo, jak to możliwe. Nie potrafię sobie wyobrazić, dlaczego <sarkazm>.
Podejście, jakie przyjęli dr Doshi i współpracownicy, polega na rygorystycznym stworzeniu zbioru danych, który jest jak najbardziej zbliżony do tego, co może być oryginałem, poprzez przeczesywanie publikacji naukowych poszczególnych firm („sponsorów”), a także stron internetowych FDA i Health Canada w poszukiwaniu wszelkich tabel lub wykazów zdarzeń niepożądanych, jakie można uzyskać z publicznych prezentacji, a następnie zebranie ich w taki sposób, aby stanowiły jak najbliższe przybliżenie „prawdziwych” danych pierwotnych, a następnie przeanalizowanie tych zbiorów danych.
Poza publikacjami w czasopismach przeszukaliśmy strony internetowe FDA (w poszukiwaniu materiałów z posiedzeń komitetów doradczych) i Health Canada (w poszukiwaniu części dokumentacji składanej przez sponsorów regulatorowi). W przypadku strony internetowej FDA wzięliśmy pod uwagę prezentacje zarówno FDA, jak i sponsorów. W każdym z tych źródeł szukaliśmy tabel z wynikami SAE, które przedstawiały informacje według określonego typu SAE; wybraliśmy najbardziej aktualną tabelę SAE odpowiadającą wymogowi FDA dotyczącemu mediany bezpieczeństwa w okresie obserwacji wynoszącej co najmniej 1 miesiąc po podaniu dawki.
SAE jest skrótem oznaczającym poważne zdarzenie niepożądane (Serious Adverse Event). Zwróć uwagę na ostatni wiersz – dwa miesiące po podaniu drugiej dawki. Z pracy Cell ze stycznia ubiegłego roku wiemy, że zarówno syntetyczne mRNA, które tak naprawdę nie jest mRNA, utrzymuje się przez co najmniej 60 dni, jak i białko spike wyprodukowane z tego mRNA, a więc „lek” jest nadal obecny przez co najmniej dwa miesiące po podaniu drugiej dawki. Prawdopodobnie byłoby o wiele lepiej, gdyby FDA nalegała, aby okres obserwacji po wystąpieniu SAE był dłuższy niż jeden miesiąc. Ale spieszyli się, bo szef sztabu Trumpa kazał im to załatwić. A więc tak to wygląda. Przyczyna i skutek.
Wracając do artykułu, w celu przeprowadzenia analiz danych dotyczących zdarzeń niepożądanych, które udało im się zebrać, Doshi i współpracownicy zastosowali listę „zdarzeń niepożądanych o szczególnym znaczeniu” (Adverse Events of Special Interest – AESI), opracowaną przez CEPI i Brighton Collaboration, a następnie zatwierdzoną przez WHO. Lista ta została opracowana przed rozpoczęciem badania. Teraz, z perspektywy czasu, mamy obszerną tabelę potencjalnych AESI firmy Pfizer, która, jak się wydaje, została opracowana PO uchwaleniu zezwolenia na stosowanie leku w nagłych wypadkach, i autorzy mogli z niej skorzystać. Jednak Doshi i współpracownicy zachowują się jak prawdziwi harcerze i zdecydowali się ocenić tylko listę AESI, która istniała przed udostępnieniem danych z badania do ich analiz, co było pozorną próbą retrospektywnego zrobienia tego, co powinno być zrobione pierwotnie.
Problem polega na tym, że nie mają oni dostępu do danych na poziomie pacjenta, więc musieli przyjąć pewne założenia dotyczące tych pierwotnych danych, zwłaszcza w odniesieniu do ich rozkładu liczbowego/statystycznego.
„Kolejnym ograniczeniem jest nasz brak dostępu do danych dotyczących poszczególnych uczestników, co zmusiło nas do zastosowania konserwatywnej korekty błędów standardowych. Obliczone 95% CI są zatem jedynie przybliżone, ponieważ nie wiemy, u których pacjentów wystąpiły wielokrotne zdarzenia. Ponadto, pomimo naszych prób usunięcia z analizy punktów końcowych skuteczności (tj. SAE oznaczonych jako COVID-19, zapalenie płuc COVID-19 i „SARS-CoV-2 test pozytywny”), nie było możliwe zidentyfikowanie i usunięcie SAE, które wystąpiły u pacjentów z poważnymi powikłaniami COVID-19 (np. ostra niewydolność oddechowa, zatrzymanie akcji serca i ostre uszkodzenie nerek), które są powszechne.”
Innymi słowy, zrobili wszystko, co w ich mocy, ale musieli przyjąć pewne założenia.
Oto tabela z kluczowymi danymi, która powstała w wyniku całej tej ciężkiej pracy:
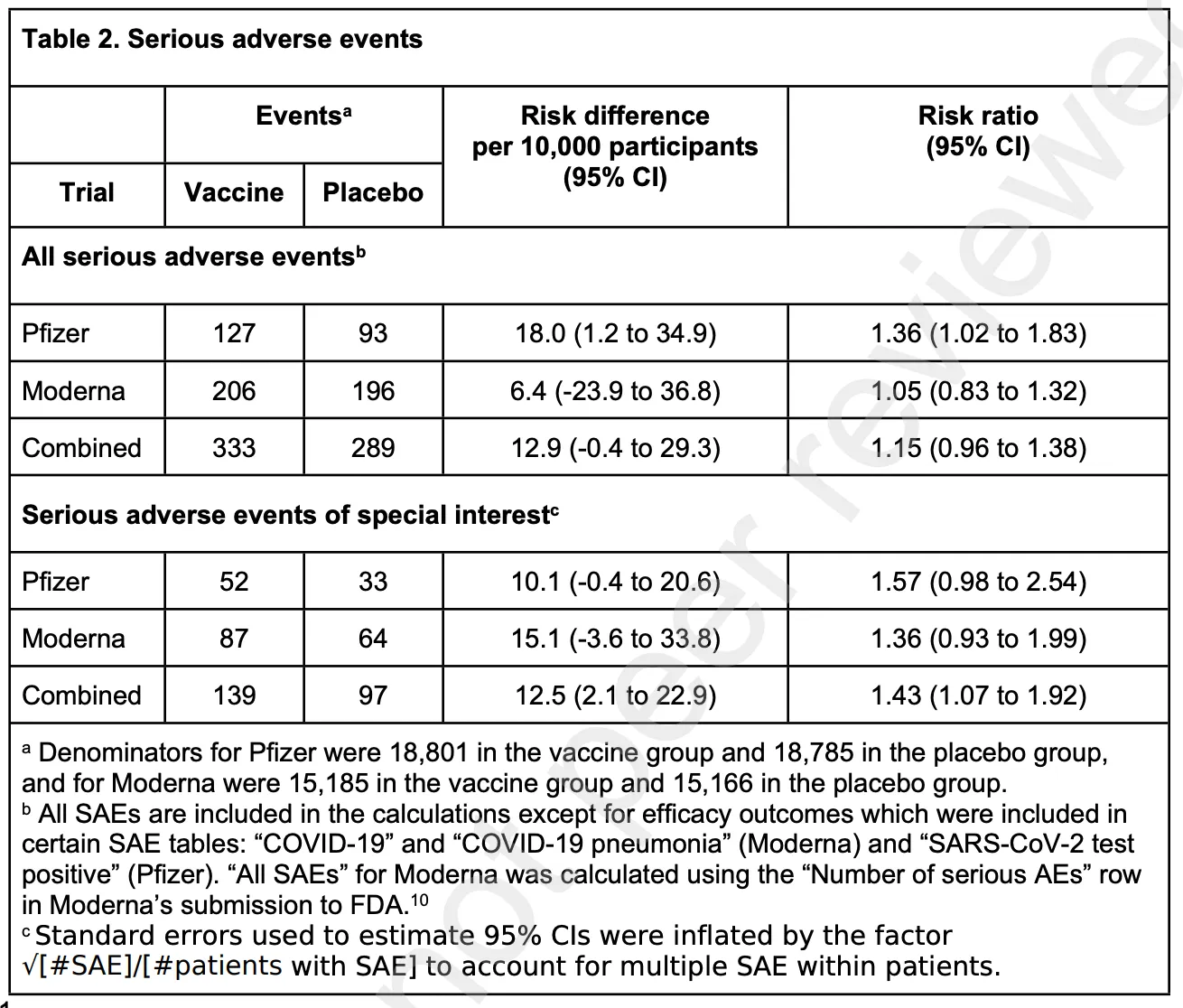
Zwróć uwagę na kolumny współczynnika ryzyka, a w szczególności na 95% przedział ufności (Confidence Interval – w skrócie CI). Współczynnik ryzyka, przy którym grupa kontrolna i grupa eksperymentalna są równoważne, wynosiłby 1,0. Większy niż 1,0 (w tym przypadku) oznaczałby, że ryzyko wystąpienia zdarzeń niepożądanych było większe u osób otrzymujących szczepionkę. Wokół tej liczby istnieje jednak pewien przedział statystyczny (przy badaniu losowym i ustaleniu progu testu statystycznego, że 95 na 100 razy wynik będzie się mieścił w tym przedziale). Tak więc jeśli przedział ufności rozciąga się od mniej niż jeden, do więcej niż jeden, nie można stwierdzić, że istnieje statystyczna różnica między wynikami w grupie kontrolnej i grupie zaszczepionej. Tak jest w przypadku wielu takich testów. Obecnie wszystkie są dość nachylone w kierunku wartości bliskich 1 i większych niż 1. Sugeruje to, że gdyby liczba badanych pacjentów była większa, wszystkie te wyniki mogłyby osiągnąć istotność statystyczną. Jest to jednak niewielka próba jak na badanie 3 fazy szczepionki. FDA pozwoliła sponsorom na takie działanie, ale takie są dostępne dane. I nie ma możliwości, abyśmy kiedykolwiek wrócili do tego punktu w czasie, ponieważ obecnie prawie wszyscy zostali zaszczepieni lub zakażeni.
W badaniach nad szczepionkami, w celu oszacowania wielkości próby badawczej, stosujemy zasadę trzech. Jeśli chcemy wiarygodnie wykryć zdarzenie niepożądane, które występuje raz na tysiąc pacjentów, powinniśmy przebadać 3000 pacjentów w grupie zaszczepionej. Tak więc w przypadku badania firmy Pfizer ma ono moc pozwalającą na wykrycie zdarzeń niepożądanych, które występują mniej więcej raz na (18 800/3) = 6 266 pacjentów. Moderna, (15 185/3) = 5 061 pacjentów. Zdarzenia niepożądane występujące rzadziej na ogół nie byłyby wykrywane na poziomie istotnym statystycznie. Korygując częstość zdarzeń niepożądanych występujących losowo w grupie kontrolnej i normalizując do liczby zdarzeń # na 10 000 pacjentów, uzyskuje się dane podsumowane w tabeli.
Należy również zauważyć, że w przypadku AESI, aby osiągnąć poziom istotności statystycznej, na który wskazują szacunki, konieczne było połączenie danych z badań klinicznych firmy Pfizer i firmy Moderna. Jest to coś, czego nigdy nie zrobiono by w warunkach „rzeczywistych”, ponieważ te dwa produkty są różne, mają różne formy podawania i są podawane w bardzo różnych dawkach mRNA.
Na podstawie powyższego można stwierdzić, że analiza ta jest prawie tak dobra, jak to tylko możliwe, biorąc pod uwagę to, z czym autorzy mieli do czynienia. Teraz jednak można również zrozumieć, dlaczego (odpowiednio) zwięźle przedstawili oni swoje wyniki („Chcemy tylko faktów, proszę pani„), a następnie wyciągnęli odpowiednio ostrożny wniosek:
„Stwierdzone w naszym badaniu nadmierne ryzyko poważnych zdarzeń niepożądanych wskazuje na potrzebę przeprowadzenia formalnych analiz szkodliwości i korzyści, zwłaszcza stratyfikowanych w zależności od ryzyka wystąpienia poważnych zdarzeń COVID-19, takich jak hospitalizacja lub zgon.”
Był to heroiczny wspólny wysiłek mający na celu próbę powrotu do punktu w historii badań klinicznych nad szczepionkami mRNA, w którym podejmowano krytyczne decyzje mające dosłownie wpływ na bieg historii. Ówczesne decyzje były podejmowane w pośpiechu, przydatność obu badań została zniweczona (celowo?) przez przedwczesne przerwanie badań i zaszczepienie grupy kontrolnej, a zebrane dane zostały w dużej mierze ukryte przed tymi, którzy chcieliby przeprowadzić niezależne analizy. Autorzy obecnej próby ponownej analizy zrobili wszystko, co w ich mocy. Jednak, jak wielokrotnie prosili dr Doshi i współpracownicy, nie można przeprowadzić właściwej analizy, dopóki nie zostaną udostępnione oryginalne zestawy danych.

Do tego czasu „zawsze będziemy mieli Paryż”. Twoje zdrówko, mała.
Zagraj to jeszcze raz, Sam. Zagraj „Jak mija czas”.
Robert Malone
Źródło: Robert Malone substack (23-06-2022)- „Pfizer and Moderna Analysis Re-do”
Przypisy tłumacza:
1. Preprint jest wstępną publikacją naukową, która nie została jeszcze w pełni zrecenzowana i nie ukazała się w tzw. renomowanym czasopiśmie naukowym. Niestety, w dzisiejszych „pandemicznych” czasach rzetelni naukowcy najczęściej nie są w stanie uzyskać możliwości publikacji w tzw. czołowych pismach naukowych, nawet swoich najbardziej solidnych prac naukowych, gdyż nałożona została cenzura i blokada na cokolwiek, co mogłoby podważyć oficjalnie obowiązującą wersję „bezpiecznych i skutecznych szczepionek”.
2. Chodzi o EUA – Emergency Use Autorization. Zezwolenie wydawane przez agencję FDA do stosowania leku w nadzwyczajnych przypadkach. Lek zakwalifikowany do stosowania w ramach EUA (lub inny produkt, np. „szczepionka przeciwko Covid-19”) stosowany jest jako lek eksperymentalny, gdyż jego skutki są niewiadomą, chociaż przypuszcza się i zakłada – bez większych dowodów naukowych, które dopiero zbiera się na podstawie obserwacji skutków tych preparatów – że pozytywne działanie przeważa nad negatywnymi skutkami. W przypadku tzw. szczepionek przeciwko Covid-19, to optymistyczne założenie okazało się całkowicie błędne.
3. Autor używa wręcz pojęcia „śmieciowym” (junk clinical trial)
4. Zobacz m.in.: Czołowe pismo medyczne wzywa FDA do natychmiastowej publikacji wszystkich danych o „szczepionkach”
5. Zobacz m.in.: Wielkie zwycięstwo sądowe: FDA zmuszone do opublikowania wszystkich dokumentów „szczepionki” w ciągu 8 miesięcy, a nie 75 lat!
Oprac. www.bibula.com
2022-06-23